2022-08-03
幹細胞是一(yī)類具有自我(wǒ)(wǒ)更新能力的多向分(fēn)化潛能細胞,在一(yī)定條件下(xià)可以分(fēn)化爲具有多種功能的組織和器官,具有重要的理論研究意義和臨床應用價值。近年來,在國家和地方政府一(yī)系列政策規範和文件指導下(xià),幹細胞轉化應用及臨床研究正在蓬勃發展,相關新藥申報工(gōng)作也在有序開(kāi)展。
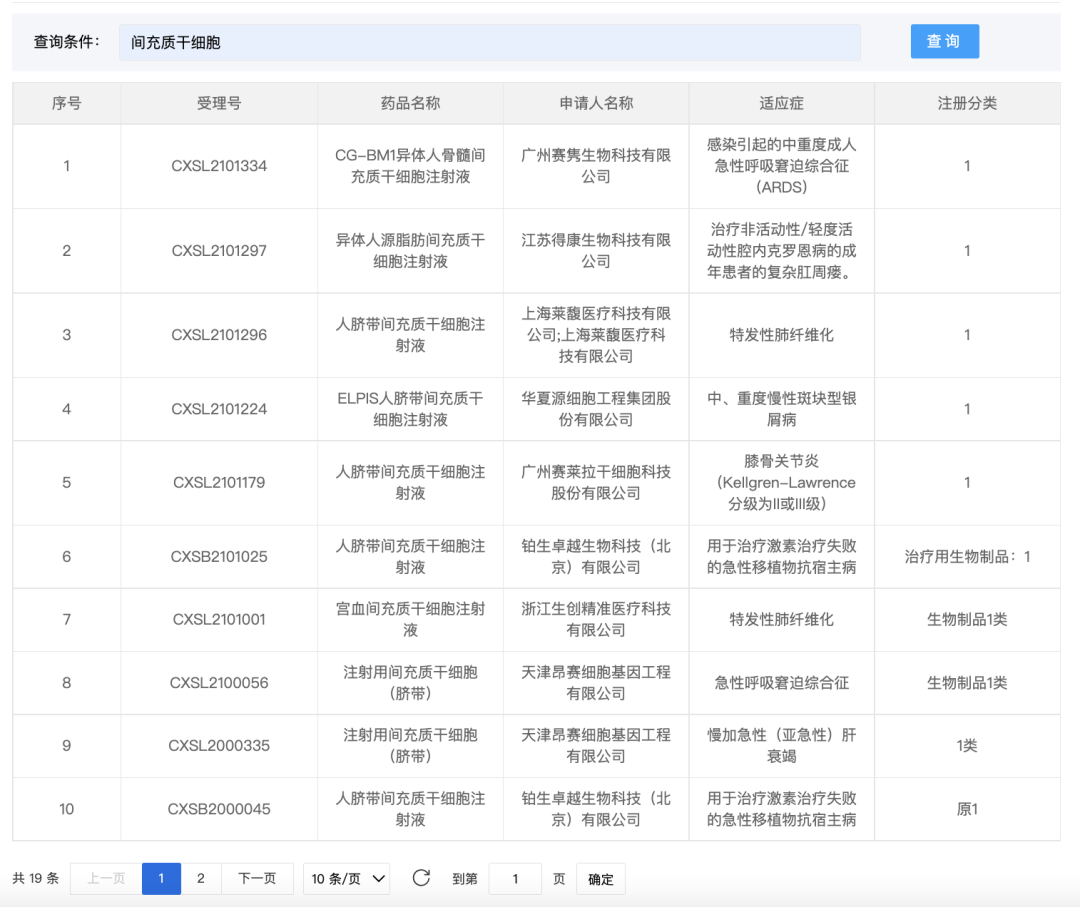
目前幹細胞通過IND審批的已有28個,其中(zhōng)間充質幹細胞19個項目,間充質祖細胞2個項目,細胞注射液4個項目,細胞自體(tǐ)回輸制劑2個項目,祖細胞1個項目,衛健委方面幹細胞備案已有112個項目。

而随着對幹細胞研究的不斷進展,諸多成果驗證了幹細胞在創傷修複、神經再生(shēng)、抵抗衰老、糖尿病、帕金森(sēn)氏症、白(bái)血病、腫瘤等疾病的治療方面有巨大(dà)潛力。其中(zhōng),幹細胞對糖尿病、腦中(zhōng)風、脊髓損傷等疾病的治療,更是已經被很多臨床研究所證實。
幹細胞在我(wǒ)(wǒ)國的發展
我(wǒ)(wǒ)國幹細胞治療的發展起步較晚,臨床研發目前以間充質幹細胞爲主,早在1993年5月衛生(shēng)部就公布了《人的體(tǐ)細胞治療及基因治療臨床研究質控要點》,2002年國家藥品監督管理局發布《藥品注冊管理辦法》,2003年國家藥品監督管理局發布《人體(tǐ)細胞治療研究和制劑質量控制技術指導原則》,2009年衛生(shēng)部發布《醫療技術臨床應用管理辦法》,明确國家建立醫療技術臨床應用準入和管理制度,對醫療技術實行分(fēn)類、分(fēn)級管理。衛生(shēng)部負責第三類高風險醫療技術的審定和臨床應用管理,幹細胞技術被劃爲第三類醫療技術。
但寬松的政策環境,除了促進了幹細胞行業的發展,但是另一(yī)方面也使幹細胞行業變得魚龍混雜(zá),良莠不齊。爲了從源頭上規範醫療技術使用、遏制濫用,2015年初,國務院決定取消“非行政許可審批”這一(yī)審批類别,在此背景下(xià),原國家衛生(shēng)計生(shēng)委取消了第三類醫療技術臨床應用準入審批。然而,取消審批并不等于放(fàng)手不管。同年,爲規範并促進我(wǒ)(wǒ)國幹細胞臨床研究,國家衛生(shēng)計生(shēng)委與食品藥品監管總局共同組織制定了《幹細胞臨床研究管理辦法(試行)》,這也是目前醫藥行業進行幹細胞臨床研究時遵循的總指導原則。

2016年後,我(wǒ)(wǒ)國政府開(kāi)始重視幹細胞行業,政府啓動幹細胞臨床研究機構和研究項目備案,促進幹細胞臨床研究健康開(kāi)展。2018年6月8日,國家藥監局新受理了幹細胞療法的臨床注冊申請,預示着我(wǒ)(wǒ)國重啓幹細胞療法在臨床上的應用。
目前,幹細胞研究及其轉化醫學已經成爲政府、科研機構和企業高度關注和大(dà)力投入的重要研究領域。
當前我(wǒ)(wǒ)國幹細胞的監管是“雙軌制”進行,一(yī)種是2015年,爲規範和促進幹細胞臨床研究,依照《中(zhōng)華人民共和國藥品管理法》、《醫療機構管理條例》等法律法規,發布《幹細胞臨床研究管理辦法(試行)》,國家衛生(shēng)計生(shēng)委與國家食品藥品監管總局負責幹細胞臨床研究政策制定和宏觀管理,根據該管理辦法,幹細胞适用于醫療機構開(kāi)展的幹細胞臨床研究。
另一(yī)種則是按照藥品進行監管。2017年發布的《藥品注冊管理辦法(修訂稿)》明确指出細胞治療類産品可以按藥品進行注冊上市,緊接着《細胞治療産品研究與評價技術指導原則(試行)》的頒布,更加明晰了細胞治療作爲藥品申報的标準。
對于兩種監管制度,出于鼓勵幹細胞産業發展的目的,無論是哪種方式在國家層面都是支持的。目前衛健委備案已經有相應的詳細政策以及方式發布,而按照藥品申報這一(yī)路徑,相關政策也在進一(yī)步完善,指導原則也更加細化。例如:2020年8月CDE頒布了公開(kāi)征求《人源性幹細胞及其衍生(shēng)細胞治療産品臨床試驗技術指導原則(征求意見稿)》,2021年8月CDE頒布了關于公開(kāi)征求《人源性幹細胞産品藥學研究與評價技術指導原則(征求意見稿)》。所以無論是哪一(yī)條路徑,法律法規都在逐步完善,都受到國家的大(dà)力支持。此外(wài),無論是選擇哪一(yī)種方式,本質上對于制劑、質量體(tǐ)系以及工(gōng)藝的要求都是一(yī)緻的。兩種方式各有趨勢,都可以得到更多的臨床數據,證明治療的可行性。但是對于大(dà)部分(fēn)藥企來說,可以成藥,進入到臨床應用,才能大(dà)規模的進行使用,使藥品走向市場,這也是最終更有利的選擇。

2018年起,爲鼓勵新藥研發和提高新藥審批效率,我(wǒ)(wǒ)國新藥申報采用新制度,新藥臨床試驗由過去(qù)的審批制度轉變爲默示許可,幹細胞作爲新藥,采用默認許可方式可爲患者争取更多的治療時間和機會。截至2月11日,目前中(zhōng)國藥品審評中(zhōng)心顯示幹細胞IND項目增至28個,幹細胞臨床轉化也獲得了實質性進展。
幹細胞申報程序
采取藥品申報路線,讓幹細胞項目走向臨床,首先要按照《幹細胞臨床研究管理辦法(試行)》和《細胞治療産品研究與評價技術指導原則(試行)》的标準,進行臨床前實驗,制備工(gōng)藝、質量控制、動物(wù)實驗、藥效、藥代以及安全性評價等。此後才能按照新藥的申報程序進行申報。
在申報前,綜述資(zī)料撰寫要注意系統全面,重點突出,綜合研究工(gōng)作,證明藥品質量穩定可靠、安全有效。具體(tǐ)資(zī)料爲綜述資(zī)料提供充分(fēn)的文獻和試驗依據,包括文獻(及翻譯稿)、試驗的實施過程,以及數據、圖表和照片等體(tǐ)現藥品研發的系統性和科學性。
CTD撰寫關鍵點



此外(wài),要注意藥物(wù)臨床試驗申請的研究資(zī)料需要可以支持開(kāi)展藥物(wù)臨床試驗,保障受試者安全。申報資(zī)料要顯示申請藥品安全性、有效性、質量可控性,經評估藥品風險不能大(dà)于獲益的。申請人也需要在規定時限内補充資(zī)料,接受藥品注冊核查、檢驗。
申請人在藥物(wù)臨床試驗申請前、藥物(wù)臨床試驗過程中(zhōng)以及藥品上市許可申請前等關鍵階段,還可以就重大(dà)問題與藥品審評中(zhōng)心等專業技術機構進行溝通交流。藥品注冊過程中(zhōng),藥品審評中(zhōng)心等專業技術機構可以根據工(gōng)作需要組織與申請人進行溝通交流。可讨論:現有研究數據是否支持拟開(kāi)展的臨床試驗、臨床試驗受試者風險是否可控等問題。
幹細胞備案程序
對于幹細胞備案,要求幹細胞臨床研究符合《藥物(wù)臨床試驗質量管理規範》。幹細胞制劑符合《幹細胞制劑質量控制及臨床前研究指導原則(試行)》。幹細胞制劑的制備符合《藥品生(shēng)産質量管理規範》(GMP)的基本原則和相關要求。

按照機構内幹細胞臨床研究立項審查程序和相關工(gōng)作制度,項目負責人須提交有關幹細胞臨床研究項目備案材料,以及幹細胞臨床研究項目倫理審查申請表。機構學術委員(yuán)會對申報的幹細胞臨床研究項目備案材料進行科學性審查。審查重點包括:
1、開(kāi)展幹細胞臨床研究的必要性;
2、研究方案的科學性;
3、研究方案的可行性;
4、主要研究人員(yuán)資(zī)質和幹細胞臨床研究培訓情況;
5、研究過程中(zhōng)可能存在的風險和防控措施;
6、幹細胞制劑制備過程的質控措施。
機構倫理委員(yuán)會将按照涉及人的生(shēng)物(wù)醫學研究倫理審查辦法相關要求,對幹細胞臨床研究項目進行獨立倫理審查。審查時,機構學術委員(yuán)會和倫理委員(yuán)會成員(yuán)将簽署保密協議及無利益沖突聲明,須有三分(fēn)之二以上成員(yuán)同意方爲有效。根據評審結果,機構學術委員(yuán)會将出具學術審查意見,機構倫理委員(yuán)會出具倫理審查批件。

機構學術委員(yuán)會和倫理委員(yuán)會審查通過的幹細胞臨床研究項目,由機構主要負責人審核立項。立項後在我(wǒ)(wǒ)國醫學研究登記備案信息系統如實登記相關信息。機構材料由省級衛計委同食品藥品監管部門審核後向國家衛計委與國家食品藥品監管總局備案。材料包括:1、機構申請備案材料誠信承諾書(shū);2、項目立項備案材料;3、機構學術委員(yuán)會審查意見;4、機構倫理委員(yuán)會審查意見;5、所需要的其他材料。
在國家的大(dà)力支持下(xià),我(wǒ)(wǒ)國有望結束幹細胞上市藥物(wù)的空白(bái),疾病的治療也會迎來更多的選擇,幹細胞研究已成爲生(shēng)命科學領域的前沿熱點,随着幹細胞治療的不斷發展,已經爲很多難治疾病的治療甚至是治愈指明了方向。相信随着臨床試驗數量不斷增加,幹細胞治療将有更光明的未來,更蓬勃的發展。
編輯:三琅

想了解更多,請關注誇克醫藥微信公衆号

地址:北(běi)京豐台區麗澤商(shāng)務區平安幸福中(zhōng)心

号")





